Prior information
A haematopoiesis precursor cell or stem cell has an ability to renew itself continuously and gradually develop to a mature cell of lymphoid or myeloid blood cell line.

In the normal blood of an adult, the largest amount of myeloid cell line representatives are neutrophilic granulocytes; there are less eosinophilic granulocytes, basophilic granulocytes, monocytes. Of lymphoid cells, a healthy adult has mature lymphocytes.
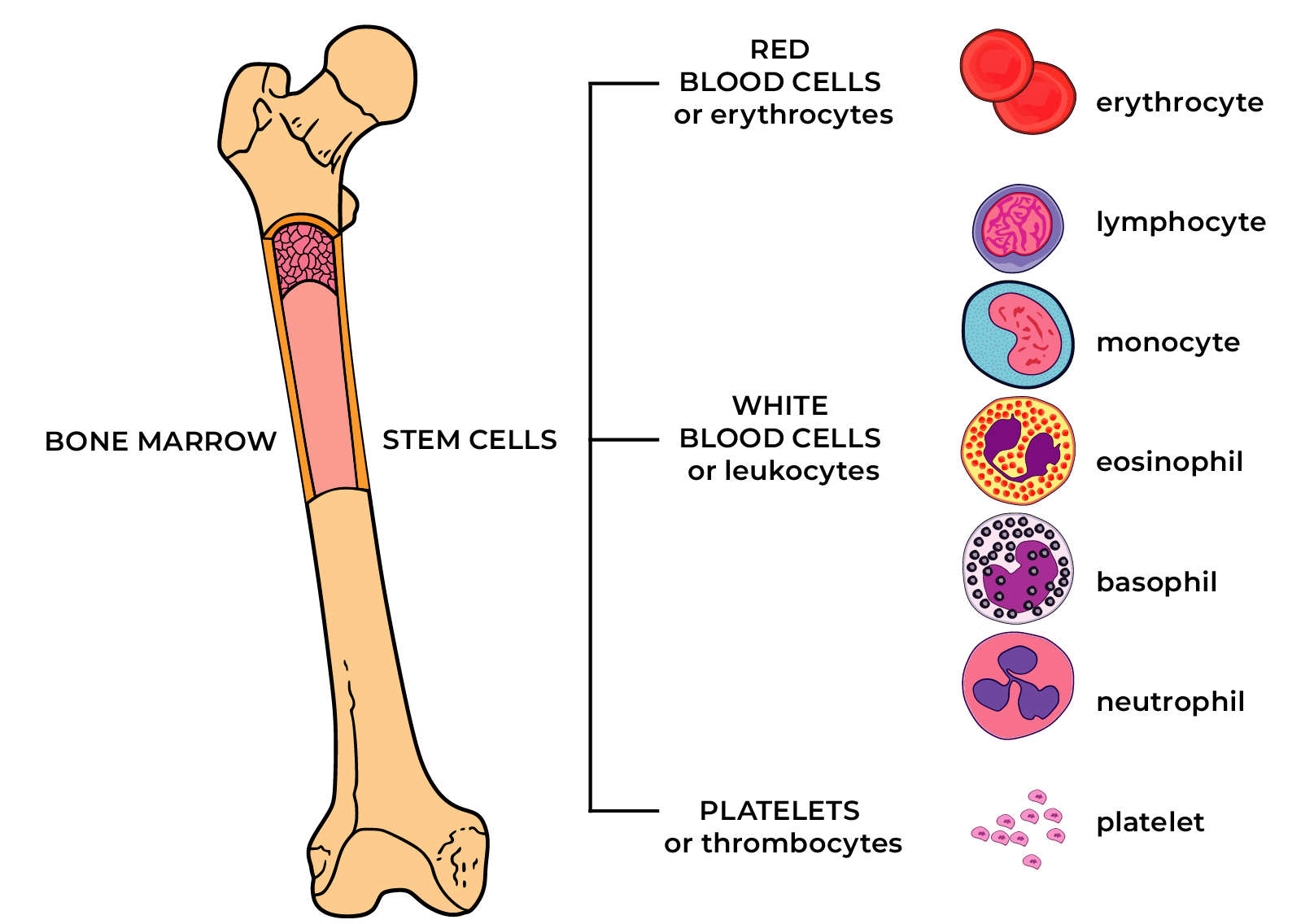
Limit values of absolute and relative counts of blood cells can be seen on the results page of a blood analysis. If you have questions, always ask for help from your doctor who is happy to interpret the results.
Depending on the origin of the cell line, haematopoietic diseases are divided into myeloid and lymphoid tumours. Both myeloid and lymphoid tumours may be divided into acute and chronic diseases (leukaemia). In the case of acute leukaemia, immature cell types called blasts dominate the blood count and bone marrow. These are diseases requiring prompt medical intervention, in which normal haematopoietic tissue is replaced with malignant tumorous cells, blasts, over a short time. Uncontrolled haemorrhages, infections, circulation disorders and impairments in the function of several organ systems can develop due to failure of normal haematopoiesis. In the case of chronic blood disorders, the tumour cells resemble regular mature blood cells. Acute leukaemia will never become chronic, but some chronic haematopoietic diseases may become aggressive diseases, for example, acute leukaemia, due to their normal course or with anti-tumour medications.
Chronic myeloproliferative tumours (internationally MPN, myeloproliferative neoplasms)
Terms and definitions
The prefix ‘myelo’ means bone marrow, ‘proliferative’ indicates rapid multiplication or proliferation of the cells. ‘Chronic’ means haematopoiesis tumour with mostly mature cells.
Chronic myeloproliferative neoplasms are diseases of the myeloid line, the main representatives are chronic myeloid leukaemia (see separate subsection for details), polycythaemia vera, primary myelofibrosis, essential thrombocythemia.
‘Polycythaemia’ means that there are more cells in the blood than normal.
‘Thrombocythemia’ means, above all, the abundance of thrombocytes or platelets.
‘Fibrosis’ means fibrous scar tissue, which, in rare cases, may oust normal haematopoietic tissue.
‘Vera’, ‘essential’ indicate tumour originating from bone marrow.
The term ‘reactive’ in the conclusion of a haematologist means that blood changes are probably caused by another disease or condition outside the bone marrow. In most cases, the causes of changes in blood counts are acute and chronic infections, other inflammatory diseases, drugs as well as smoking, alcohol.
Epidemiology
All myeloproliferative neoplasms (MPN) are rare or very rare diseases. For example, the incidence of polycythaemia vera or essential thrombocythemia may be around two new cases per 100,000 persons per year, primary myelofibrosis is even rarer – 0.22-0.5 new cases per 100,000 persons per year. For comparison, type II diabetes is diagnosed in Europe with an incidence of around seven new cases per 1000 persons per year. Polycythaemia vera is found more often in people over 50; in men a little more frequently than in women. Patients becoming ill with essential thrombocythemia are more often over 60 (men and women equally), but around 20% are less than 40 years old at diagnosis (women more often).
Causes of MPN
In most patients with polycythaemia vera, a change in the Janus-kinase 2 gene (abbreviation JAK2) is found, therefore the production and destruction of blood cells is no longer subject to the body’s normal regulation. The life-span of blood cells is prolonged, their total volume in the bone marrow and blood increases with time.
This change in JAK2 is most often present in patients with essential thrombocytopaenia and primary myelofibrosis. Less often, changes in calreticulin (CALR) and MPL genes occur in patients with essential thrombocytopaenia and primary myelofibrosis.
These changes develop during the course of life and are not hereditary.
The causes of gene mutations are not completely known or avoidable.
MPN diagnosis
MPN suspicion is in most cases based on blood count. In such cases, a haematologist orders an additional blood analysis for molecular genetic investigation. A change in the JAK2, CALR or MPL gene gives a clearer suggestion that it could be a disease of the MPN group. Regrettably, it is not always possible to determine which disease of the MPN group it is on the basis of blood analyses and genetic tests. For diagnosis elaboration and especially for evaluation of haematopoiesis status before initiating specific anti-tumour treatment, a bone marrow investigation is required.
The risks and prognosis of MPN group diseases differ in patients of different ages and with different diagnoses.
In polycythaemia vera (PV), the number of all blood cells, especially red blood cells, increases. Haemoglobin (over 165) and haematocrit (over 49%, indicates the ratio of blood cells and blood plasma) are especially high.
Main disease signs:
- reddish face, whites of eyes, hands and feet
- in ca 40%, skin itching, especially after exposure to warm water
- headache, drowsiness, tiredness
- sometimes changes are discovered associated with acute cardiovascular disease (for example, after myocardial infarction or stroke)
- many patients don’t complain of anything; the disease is found accidentally
Essentsiaalse trombotsüteemia (ET) korral suureneb trombotsüütide hulk veres ≥ 450 x 109/l
In essential thrombocytopaenia (ET) platelet count in the blood increases to ≥450 x 109/L.
- in many patients, thrombocytosis is found accidentally
- sometimes blood changes are discovered in association with an acute disease with thrombosis or blood vessel obstruction
- sometimes haemorrhages occur, especially when the platelet count is >1000 x 109/L
- in 30-50%, spleen enlarges (may cause feeling of inconvenience or fullness)
The biggest health risks of PV and ET are associated with arterial and venous thrombosis. The risk of thrombosis increases for those aged over 60 and with concurrent diseases such as type II diabetes and hypertension.
In myelofibrosis (MF), normal haematopoiesis regulation is impaired due to genetic changes. An abnormal amount of fibrous scar tissue or fibrosis occurs in the bone marrow. Myelofibrosis may gradually develop in the background of other bone marrow diseases (for example, polycythaemia vera, essential thrombocythemia). Myelofibrosis can also develop independently – in such a case, the tumour is called primary myelofibrosis. The spleen and liver usually enlarge, causing a compensatory take over of the production of blood cells.
Main disease signs:
- number of blood cells decreases with time
- due to anaemia (low haemoglobin) tiredness, weakness, palpitations, decrease in physical performance
- frequent infections due to shortage of leukocytes or white blood cells
- haemorrhages due to shortage of thrombocytes or platelets
- feeling of full stomach, abdominal pain, abdominal gases due to enlargement of spleen (and also liver)
- general symptoms – increased nocturnal sweating, weight loss, tiredness, fever
- bone and joint pain, leg cramps
The main objective of PV and ET treatment is the reduction of thrombosis risk.
Treatment options may include:
- low-dose aspirin (so-called baby aspirin);
- bloodletting or therapeutic phlebotomy to keep haematocrit <45%;
- cytoreductive agents to inhibit cell growth; or
- uric acid reducing agents (if required).
The objective of MF treatment is the alleviation of symptoms and the improvement of quality of life.
Treatment choices may include the following options depending on disease load and risks:
- in the case of anaemia, transfusions of red blood cells, erythropoietin, hormonal treatment with glucocorticoids
- if required, removal of excess iron from the body (iron accumulates in connection with blood transfusions)
- reduction of tumour load: chemotherapeutic agents (e.g. hydroxycarbamide or Hydrea), JAK-inhibitors
- uric acid-reducing agents
- rarely in younger patients, transplantation of stem cells from other people (allogeneic)
What can be done by the patient themselves?
MPN group diseases cannot be prevented, but with self-preserving behaviour, it is possible to reduce the risks of complications.
Main recommendations for adaptation with the disease:
- avoid dehydration
- don’t smoke
- eat a healthy, balanced diet
- avoid taking iron-containing food supplements or drugs (please consult beforehand with a haematologist!)
- maintain a normal weight and practice recreational sports, if possible
- avoid prolonged stress position, e.g. sitting
- keep blood pressure, cholesterol and blood glucose under control
- consult with a haematologist when planning pregnancy and getting pregnant
- vaccination against for example COVID-19, influenza is allowed and very necessary!
Compiled by: Dr Halliki Kõdar
Last updated: May 2021